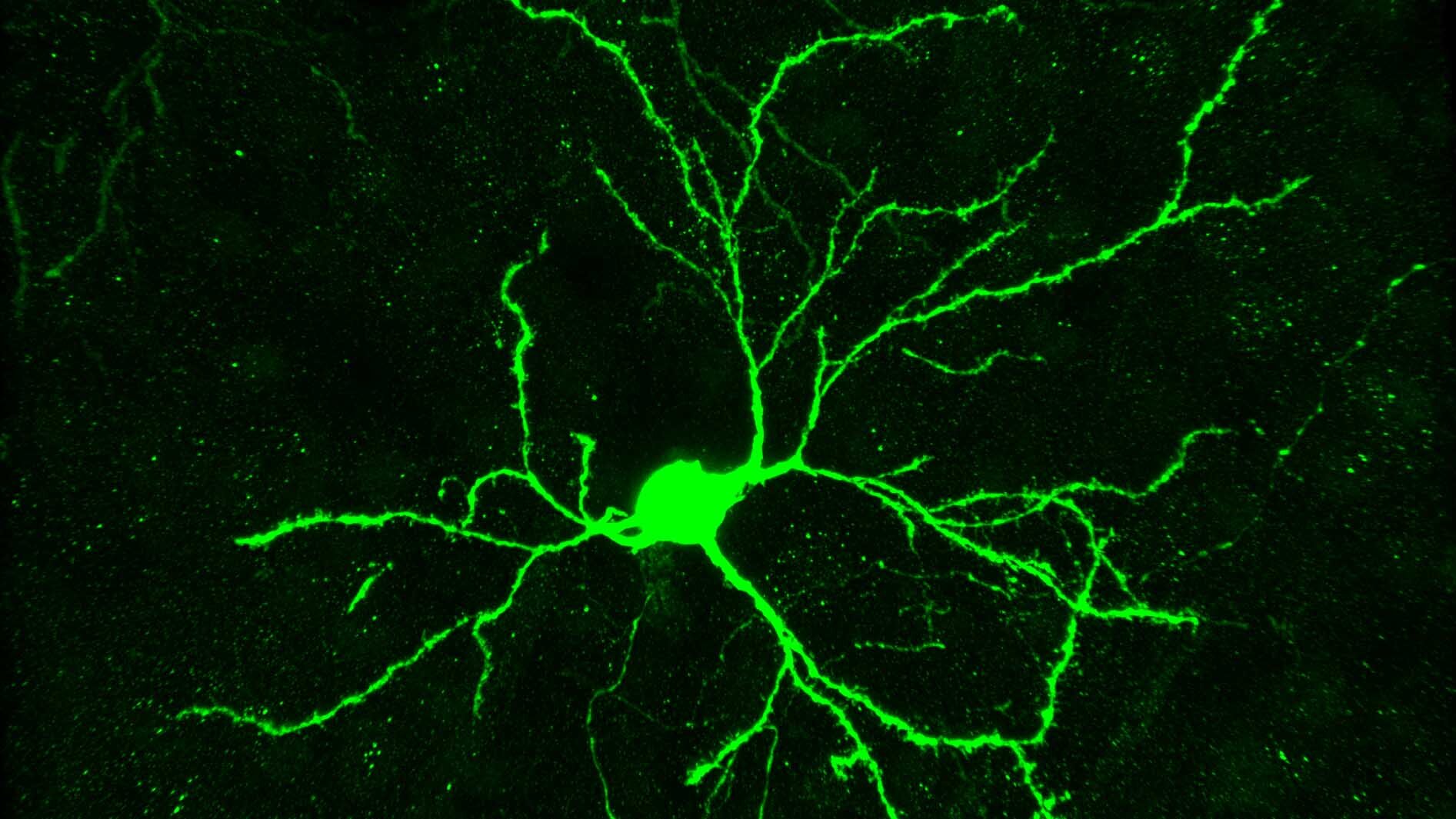
Laboratory of Synaptic Proteostasis
Our lab studies proteins and focuses on how failures in protein quality control disrupt synaptic function, contribute to neurodegeneration, and accelerate the aging process.
The role of impaired synaptic vesicle machinery proteostasis in Alzheimer’s disease pathogenesis. This project examines how impaired presynaptic proteostasis disrupts the synaptic vesicle (SV) cycle and drives early synaptic dysfunction in Alzheimer’s disease (AD), and whether targeting SV machinery can prevent or slow prodromal progression. Synapse loss is an early hallmark of AD that correlates strongly with dementia severity, yet the mechanisms that initiate presynaptic failure remain incompletely defined. Our recent work shows a selective impairment of protein turnover in axon terminals, leading to the accumulation of old presynaptic SV-associated proteins and an enlarged SV pool that precedes overt synapse loss in App knock-in mouse models and brains from individuals with Down syndrome. We further found that the FDA-approved drug levetiracetam reverses presynaptic protein buildup, restores non-amyloidogenic APP processing, and that these effects require the SV protein SV2A. Building on these findings, we are investigating whether modulating synaptic vesicle endocytosis with genetic and pharmacological approaches can limit amyloidogenic APP processing and thereby reduce Aβ accumulation.
Tau in presynaptic dysfunction during the cellular phase of AD. Using metabolic stable-isotope labeling in transgenic P301S Mapt mice combined with mass spectrometry, this project maps when and where tau becomes phosphorylated as tau pathology emerges and progresses. We then identify synaptic and non-synaptic proteins whose turnover is disrupted alongside tau changes, factors that may promote paired helical filament formation and neurofibrillary tangles or contribute to neuronal dysfunction and death. A key goal is to define tau’s role in early amyloid-driven presynaptic dysfunction and to test whether antisense oligonucleotide (ASO)–mediated tau reduction can limit tau pathology and weaken amyloid–tau crosstalk. Together, these studies connect tau modification, proteostasis failure, and synaptic decline, while evaluating tau lowering as a therapeutic strategy for tauopathies.
The role of long-lived proteins in development and aging. This project aims to accelerate our understanding of the biological significance of long-lived proteins (LLPs) with lifetimes measured in months and, in some cases, persisting for as long as the organism itself. We have identified LLP populations across multiple organ systems, including the brain, heart, inner ear, and the female reproductive system, highlighting LLPs as a widespread and potentially fundamental feature of mammalian biology. LLPs can be both vital and vulnerable: they provide durable structural and functional stability within long-lived cellular architectures (for example, nucleoporins, ATP synthase complexes, and histones), yet their exceptional longevity also increases the likelihood of cumulative damage, creating points of susceptibility that may contribute to aging and disease. While prior studies have offered important snapshots of protein lifetimes, they have not captured LLP dynamics across development, tissue context, and aging. Our objective is to leverage LLPs as biological “time capsules” to reveal new principles of developmental biology, identify vulnerabilities that emerge with aging, and map pathways of impaired proteostasis that contribute to neurodegeneration.
Enhancing cochlear proteome fidelity to prevent noise-induced hearing loss. The goal of this project is to develop a novel, targeted therapeutic to prevent noise induced hearing loss. The proposed research strategy is based on our finding that noise exposures causing synapse loss, severely impair the cochlear proteome and active the proteostasis network. Our recent results revealed that acoustic trauma activates and overwhelms the ubiquitin proteasome system in spiral ganglion neurons. Taken together with the previous finding that loud noise activates the heat shock response, it is increasingly clear that altered protein quality control plays a central role in the cochlea’s response to auditory insults. The rational is that small molecule regulators of the proteostasis network can protect the cochlear proteome and prevent synapse loss through the activation of stress-responsive signaling.